 On July 28, 2014, the U.S. Food and Drug Administration (FDA) issued new guidelines regarding the administration of the agency’s 510(k) program for providing quick approvals of updated medical devices. The new guidance from the FDA is believed by some to be a response to criticisms regarding alleged abuses of the program that may have allowed defective medical devices to be used without adequate testing beforehand.
On July 28, 2014, the U.S. Food and Drug Administration (FDA) issued new guidelines regarding the administration of the agency’s 510(k) program for providing quick approvals of updated medical devices. The new guidance from the FDA is believed by some to be a response to criticisms regarding alleged abuses of the program that may have allowed defective medical devices to be used without adequate testing beforehand.
The program was first introduced in the 1980s to “fast-track” the approval of simpler items like tongue suppressors and Band-Aids. Manufacturers could assert that updated versions of such products were “substantially equivalent” to previous items, and this could lead to avoidance of extensive testing that the FDA typically requires.
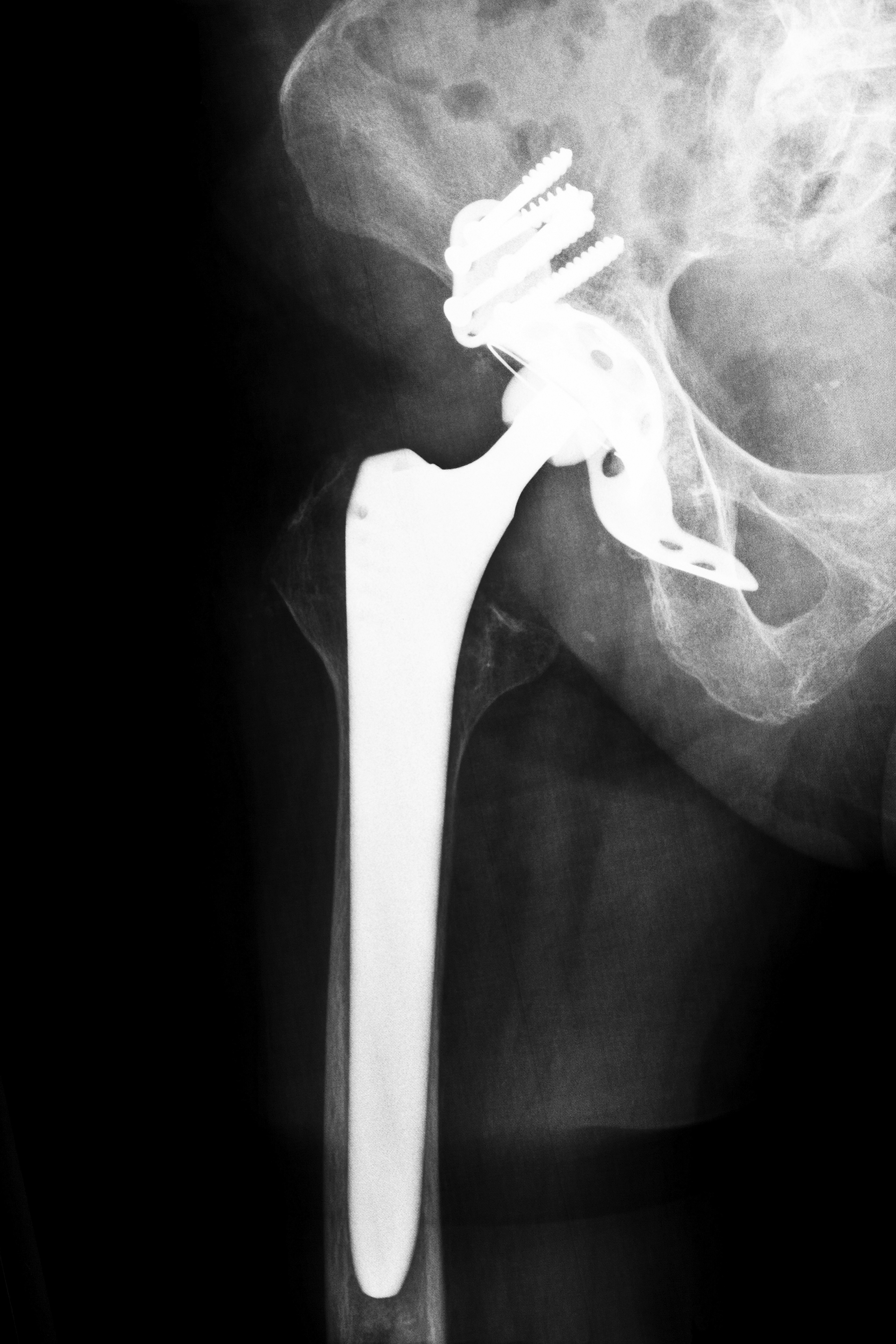
However, over the years, manufacturers have submitted more complex devices for approval under the 510(k) program. This led, critics claim, to defective medical devices like certain defibrillators and artificial hip replacements being used in patients. Subsequent litigation has sometimes proven that defective medical devices that were fast-tracked through the 510(k) program did in fact harm certain patients in Louisiana and elsewhere.
In 2011, Chairman Dr. David Challoner of the Institute of Medicine expressed the view that the 510(k) program should be disbanded altogether. He asserted that, too often, medical device manufacturers had persuaded the FDA to approve inadequately tested, defective medical devices via the program. Dr. Challoner suggested that a number of devices had been proven to be harmful only after they were implanted in or used on many patients. In Louisiana and elsewhere across the country, lawsuits have been filed on behalf of patients who were allegedly injured by defective devices.
The updated guidelines that the FDA put into effect on July 28, 2014, seek to tighten the requirements for 510(k) approvals. Substantial equivalency can only be claimed when 1) a newer medical device is used in the same fashion that the predicate device was used, and 2) the technological qualities of the updated device are the same as the previous device possessed. When there are in fact technological changes, a manufacturer has to clearly address safety concerns that these changes may bring about. Then, scientific information, clinical trials or other data must be submitted to properly support the manufacturer’s claims.
Despite the issuance of the newer FDA guidelines, allegedly defective medical devices have been used in a variety of medical procedures in Louisiana and across the nation. The use of some devices, such as certain kinds of artificial hip replacements and some types of transvaginal surgical mesh, has resulted in compensation being paid to affected individuals.
Have You Been Injured by a Defective Medical Device?
If you believe that a defective medical device has harmed you or a loved one, Attorney Group for Louisiana is available to answer your questions. We can offer you a complimentary consultation and also connect you with an affiliated attorney if you decide to pursue a claim.

{kind=link}