 A much-debated system for quick approvals of new medical devices is the subject of modified guidelines issued by the Food and Drug Administration (FDA). The long-established 501(k) program was abused by medical device manufacturers, according to some in Oklahoma and throughout the country. As a result, it is claimed, dangerous devices were distributed and used without appropriate testing.
A much-debated system for quick approvals of new medical devices is the subject of modified guidelines issued by the Food and Drug Administration (FDA). The long-established 501(k) program was abused by medical device manufacturers, according to some in Oklahoma and throughout the country. As a result, it is claimed, dangerous devices were distributed and used without appropriate testing.
On July 28, 2014, the Food and Drug Administration issued updated rules and procedures regarding a key provision in the 501(k) premarket approval program. This update to the medical device approval process was communicated to those in the FDA as well as to those in the medical device industry.
Specifically, the July 28 update clarifies how the FDA determines if a given medical device is considered to be “substantially equivalent” to a device that was previously approved. This provision has made it possible for manufacturers to bypass usual clinical trials that must occur to get an initial FDA approval for a given device.
Now, the FDA has published a guide that identifies risk-benefit factors that must be considered when the “substantially equivalent” claim is being asserted by a manufacturer.
Critics have assailed the past program, stating that, since the 1980s, the phrase “substantially equivalent” has been rendered virtually meaningless. Because of the abuse of the “substantially equivalent” claim, the public in Oklahoma and elsewhere has often been led to believe that new medical devices were adequately tested when in fact they were not.
Medical device manufacturers have routinely claimed that new devices are nearly identical to those previously approved. However, in many cases, they have gone on to market these newer devices by claiming that they featured “revolutionary” or otherwise improved designs and/or materials. Essentially, certain manufacturers have tried to have it both ways, downplaying the changes with the FDA while simultaneously promoting the changes with the public and the medical community, in Oklahoma and across the United States.
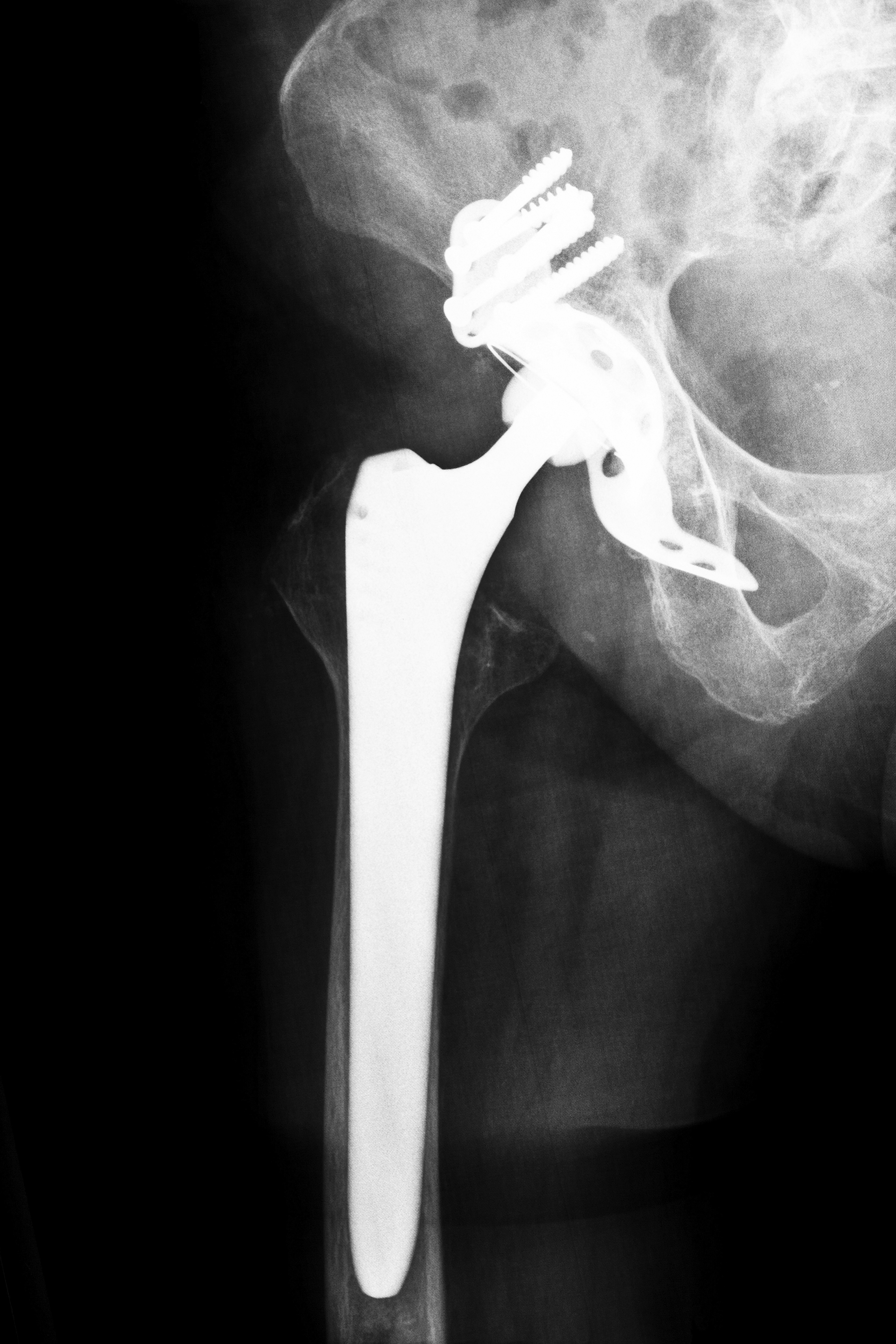
The FDA originally established the “substantially equivalent” standard to speed approval of items like tongue suppressors and Band-Aids. Gradually, the program expanded to include artificial joints, defibrillators, stents and surgical mesh. In a number of cases, these devices were approved, only to be subsequently recalled because they were claimed to be defective medical devices.
Injured by a Defective Medical Device?
If you, a family member or someone you know has been adversely affected by a defective medical device such as an artificial hip joint or transvaginal surgical mesh, please contact us at Attorney Group for Oklahoma. We can schedule a complimentary consultation to discuss your case and help you determine if you have a claim. If you decide to pursue your claim, we can connect you with an affiliated attorney who can assist you through the legal process. Contact us today to learn more.

{kind=link}