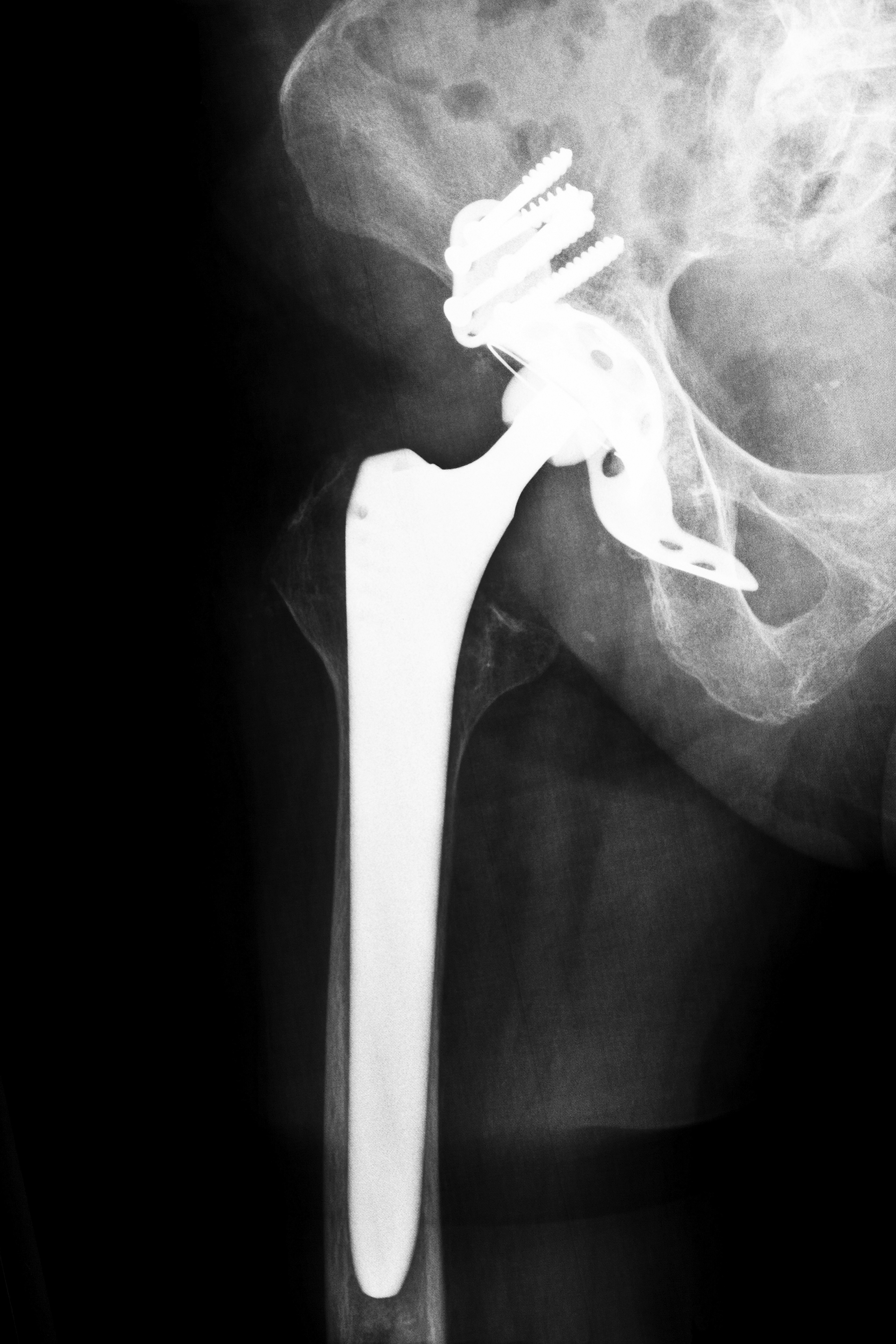
 The Food and Drug Administration (FDA) has updated approval guidelines for medical devices after lobbyists for the $350 billion industry, pressured them. Manufacturers argue that the United States is falling behind other countries that do not have similar approval rules when it comes to new devices. Safety experts say the updates are dangerous to the public as evidenced in a 2011 report by the Institute of Medicine released which found the current process was seriously flawed. In fact, the report indicated that failure to properly approve some devices led to a higher than normal number of defective medical devices it in Florida and other states.
The Food and Drug Administration (FDA) has updated approval guidelines for medical devices after lobbyists for the $350 billion industry, pressured them. Manufacturers argue that the United States is falling behind other countries that do not have similar approval rules when it comes to new devices. Safety experts say the updates are dangerous to the public as evidenced in a 2011 report by the Institute of Medicine released which found the current process was seriously flawed. In fact, the report indicated that failure to properly approve some devices led to a higher than normal number of defective medical devices it in Florida and other states.
Proposed Changes
The Food and Drug Administration Safety and Innovation Act, which the Senate passed in 2012, was designed to speed up the release of medical devices. In doing so, the act failed to address problems that have already allowed defective medical devices to be used on patients. Medical devices with known safety issues, even if they have been recalled, may be used as the basis for new devices. There is still no device problem national registry. The FDA may still not require post-market studies. Safety experts in Florida and other states say that these measures would not have slowed the process significantly, but would have provided better safety measures for consumers.
Approval Methods
The problem lies in one of the two approval methods used by the FDA for devices. The 510(k) process is used for approximately 95 percent of devices designed to treat patients. This process relies on demonstrations by manufacturers that the product is similar to devices that have already been approved and are on the market. In many cases, the devices are used for two completely different medical processes, such as testing for tumors and testing for drugs. The second method for approval, which is reserved for potentially high-risk devices, requires more testing, but it still fails to ensure that devices are safe. In fact, prescription drug approval is far more thorough than medical device approval
Delayed Action
Experts claim that an accelerated approval process is not the only issue facing consumers regarding defective medical devices. In many cases, the FDA is slow to respond when problems are reported to them about defective medical devices. One Florida case is evidence that a slow response by both a manufacturer and the FDA led to one woman’s injury. The plaintiff had a Trident hip replacement device implanted six days after the manufacturer was notified that the replacement failed to comply with federal standards. The hip replacement failed, and the woman was required to have additional surgeries. Had both Stryker and the FDA acted more quickly, the woman may not have suffered after her implant.
Injured by a Defective Medical Device?
Unfortunately, with the new updates to approval guidelines, defective medical devices may injure more people. If you or a loved one have been injured by a defective medical device, contact Attorney Group for Florida for a free consultation regarding a personal injury case. We can help answer your questions and connect you with an affiliated attorney. Contact us today to learn more.

{kind=link}